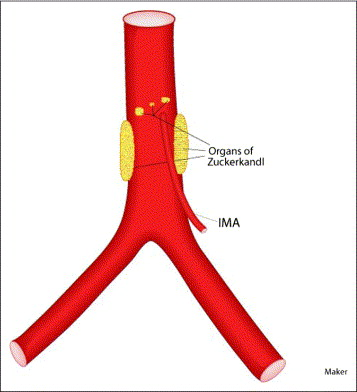
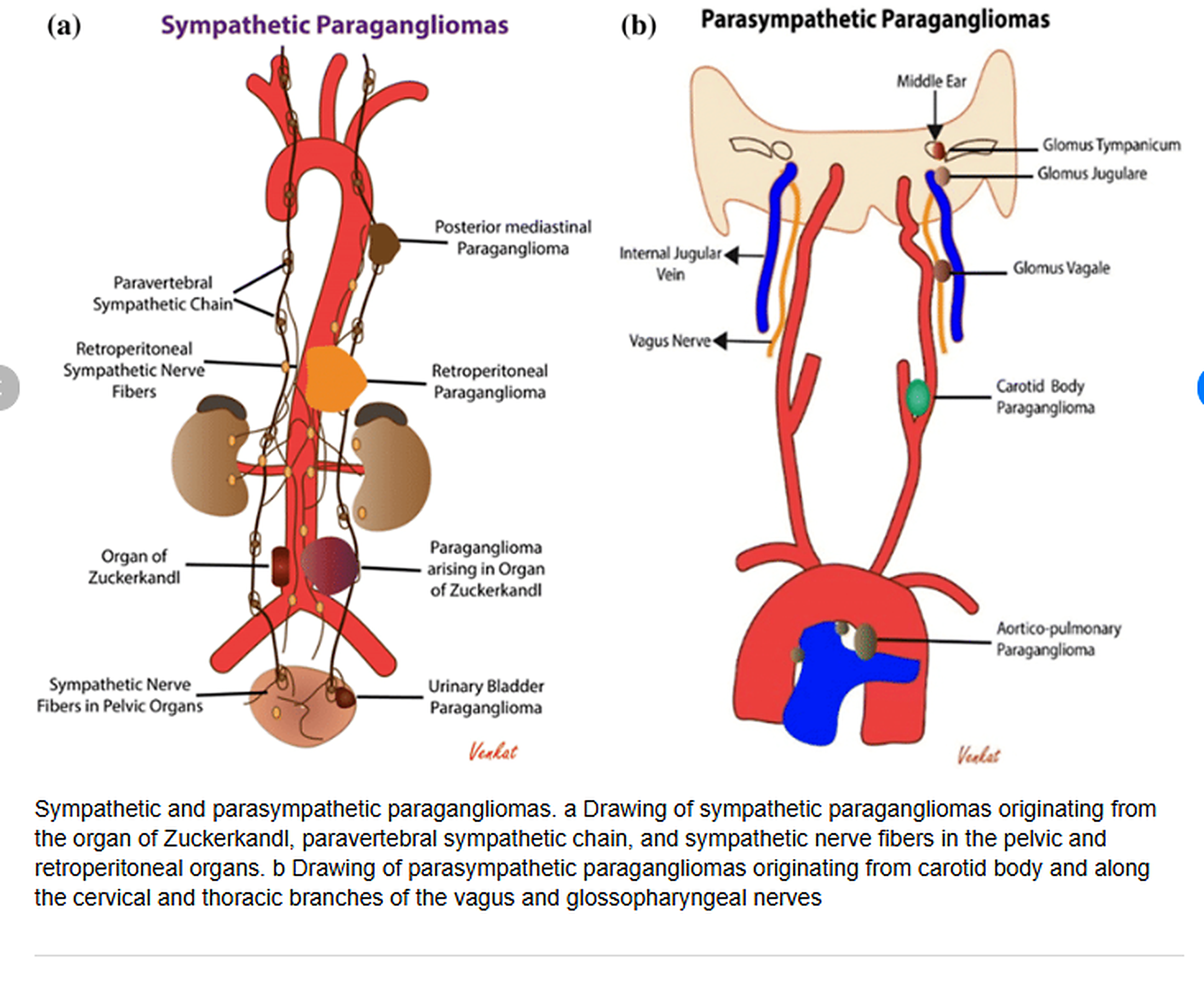
Organ of Zuckerkandl (Pheochromocytoma)
Key Images
Look For First
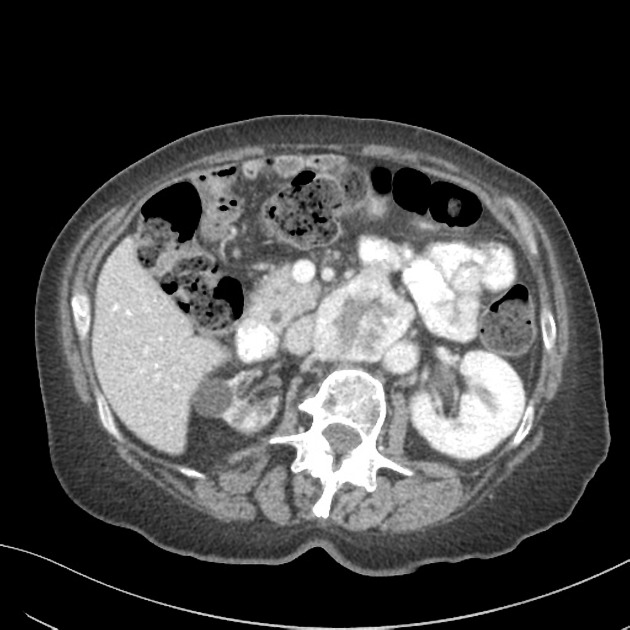
- Small to medium-sized mass located along the infrarenal aorta, typically at the level of the inferior mesenteric artery origin
- Intense enhancement on arterial phase imaging, reflecting high vascularity from catecholamine-secreting tissue
- Mass positioned between the aortic bifurcation cranially and extending to the level of the superior mesenteric or renal arteries
Key Image Findings
- The organ of Zuckerkandl is a collection of chromaffin cells derived from neural crest located along the aorta, typically at the origin of the inferior mesenteric artery where the highest concentration normally resides.
- On contrast-enhanced CT or MRI, pheochromocytomas arising from the organ of Zuckerkandl demonstrate avid arterial phase enhancement due to their hypervascular nature from rich catecholamine-secreting tissue.
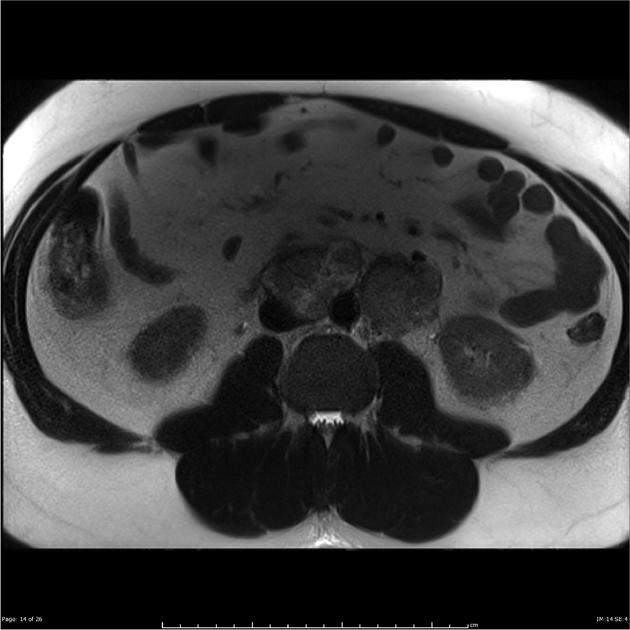
- MRI typically shows T2 hyperintense signal intensity within the mass, reflecting the cellular composition of paragangliomas and pheochromocytomas.
- The mass is characteristically located in the retroperitoneal space along the infrarenal aorta, typically between the renal arteries superiorly and the aortic bifurcation inferiorly.
- Delayed or portal venous phase imaging may show continued enhancement or slight washout pattern depending on the degree of fibrosis and cellularity within the tumor.
- Associated complications such as aortic displacement or compression may be evident on imaging when the tumor reaches significant size.
- Functional imaging with MIBG scintigraphy or PET imaging (using fluorodeoxyglucose or dopamine analogues) can confirm the catecholamine-secreting nature of the lesion.
Differential Diagnosis
- Neuroblastoma: typically occurs in children, often presents as a larger infiltrative mass with greater heterogeneity and calcification; however, rare involvement of the organ of Zuckerkandl has been reported.
- Lymph node metastasis: lacks the characteristic enhancement pattern and T2 hyperintensity of paraganglioma; lymph nodes are typically smaller and multiple.
- Aortic aneurysm or pseudoaneurysm: shows flow artifact or heterogeneous luminal enhancement rather than solid mass enhancement; location is within the aortic wall rather than adjacent tissue.
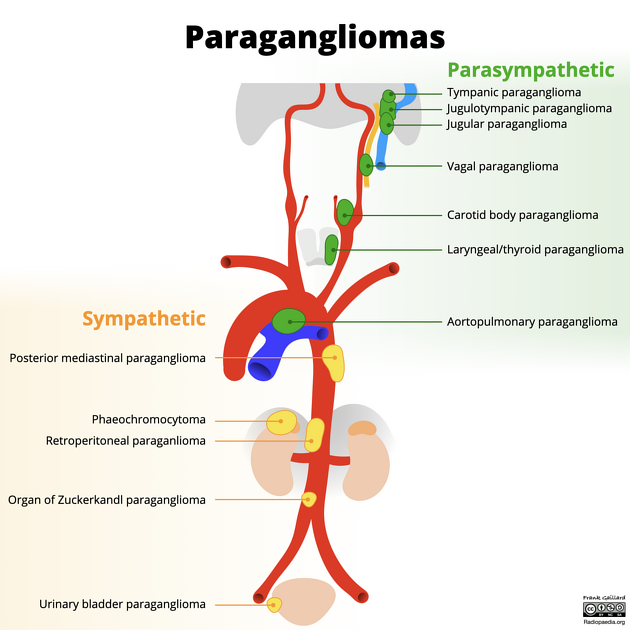
- Adrenal pheochromocytoma: arises from the adrenal medulla located at the superior pole of the kidney rather than along the infrarenal aorta; key distinguishing feature is anatomic location.
- Caval or aortic thrombosis: demonstrates different enhancement pattern with lack of solid mass characteristics and presence of flow-limiting clot.
- Retroperitoneal sarcoma: typically larger at presentation, less intensely enhancing, and lacks the catecholamine secretion symptoms that prompt imaging of the organ of Zuckerkandl.
Discussion
The organ of Zuckerkandl is a normal embryologic remnant of chromaffin tissue that regresses after birth; however, pathologic transformation to pheochromocytoma or paraganglioma can occur in a small percentage of cases.
Pheochromocytomas arising from the organ of Zuckerkandl are extra-adrenal paragangliomas that can produce catecholamines, causing hypertension, palpitations, and sweating similar to adrenal pheochromocytomas.
The infrarenal aortic location of the organ of Zuckerkandl is a key diagnostic clue that distinguishes these tumors from adrenal pheochromocytomas and aids in preoperative localization for surgical planning.
Functional testing with plasma free metanephrines or 24-hour urinary metanephrines should be performed in all patients with suspected pheochromocytoma arising from the organ of Zuckerkandl to confirm catecholamine excess.
MIBG scintigraphy or PET imaging can provide functional confirmation of catecholamine secretion and detect multifocal disease, which occurs in approximately 10-15% of pheochromocytomas.
Genetic testing and screening for associated syndromes (VHL, NF1, SDH mutations) should be considered, as extra-adrenal pheochromocytomas have a higher association with hereditary syndromes compared to adrenal tumors.
Reporting Pearls
Describe the mass by its relationship to the aorta and inferior mesenteric artery: 'A small enhancing mass is identified in the retroperitoneum along the infrarenal aorta at the level of the inferior mesenteric artery origin, consistent with the organ of Zuckerkandl; given its intense arterial enhancement and T2 hyperintensity, pheochromocytoma or paraganglioma should be considered in the appropriate clinical context.'
Pitfalls
- Failure to recognize the characteristic infrarenal aortic location can lead to misdiagnosis as an adrenal lesion or other retroperitoneal mass, delaying appropriate clinical and surgical management.
- Assuming all retroperitoneal masses along the aorta are adenopathy or other benign processes without considering paraganglioma; the intense enhancement and location should raise suspicion for neuroendocrine pathology.
- Overlooking the need for functional imaging confirmation with MIBG or PET in patients with anatomically suspicious lesions, potentially missing the diagnosis or failing to detect metastatic disease.
- Not obtaining adequate arterial and delayed phase imaging, which may be necessary to characterize the enhancement pattern and differentiate from vascular structures or thrombus.