Pheochromocytoma
Key Images
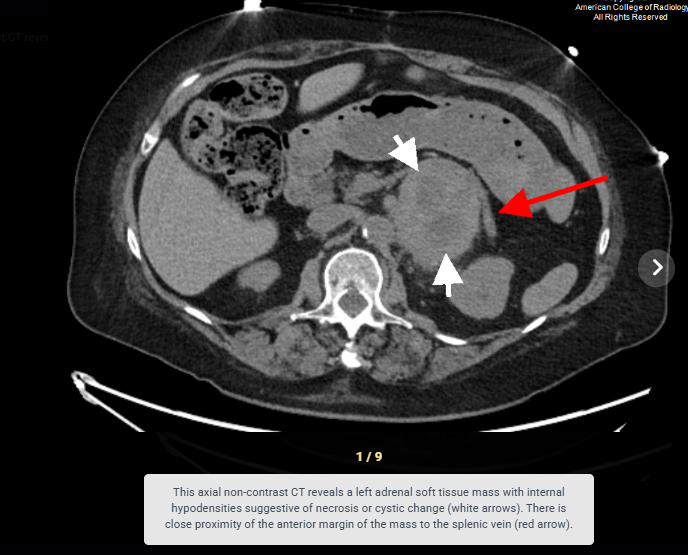
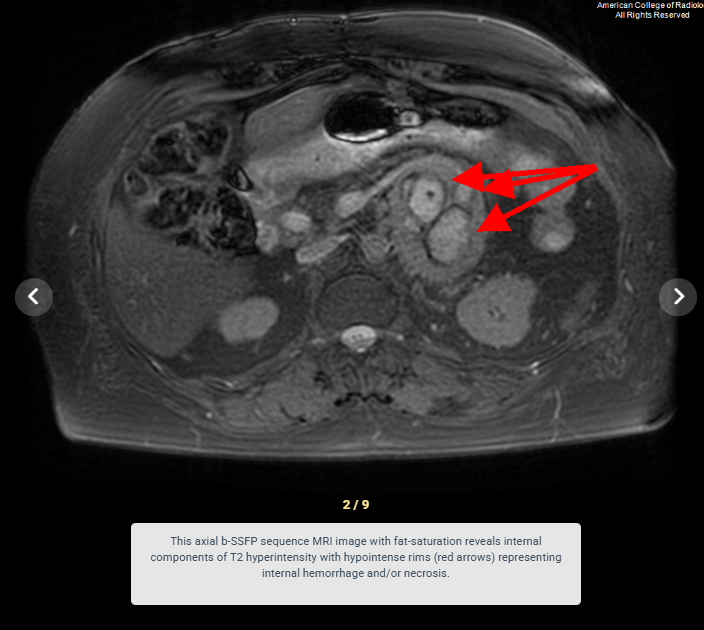
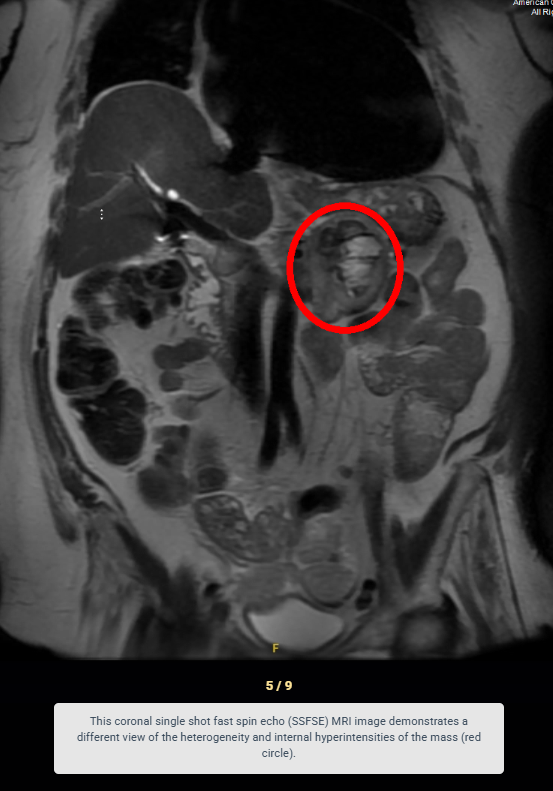
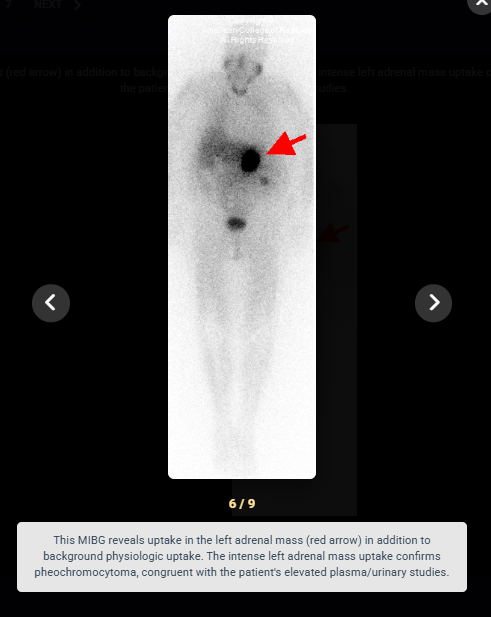
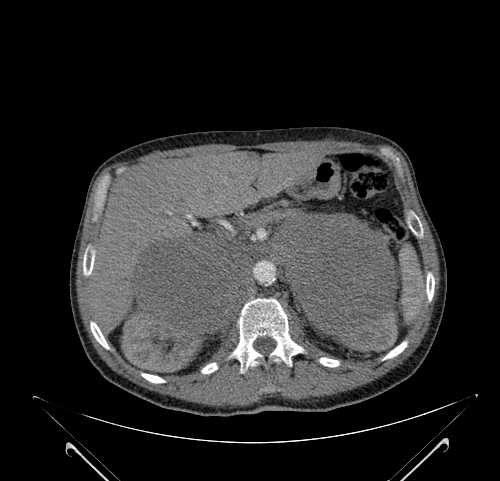
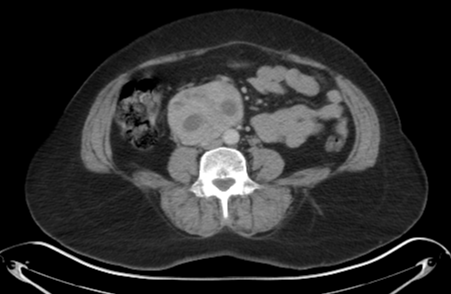
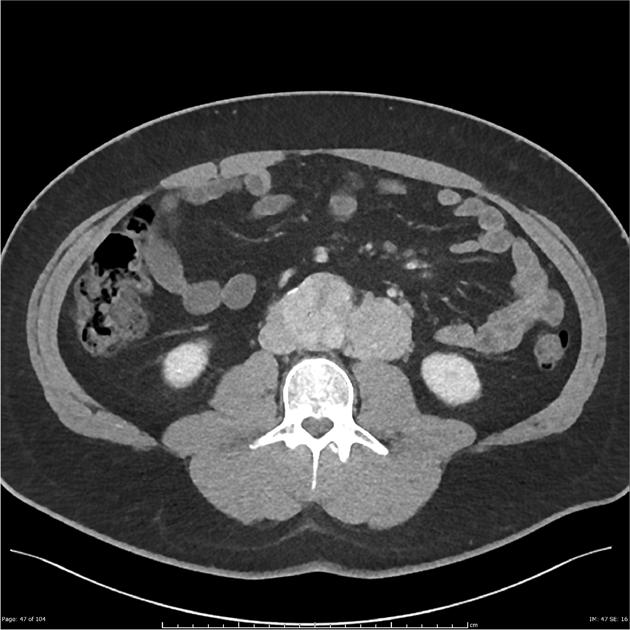
Look For First
- Marked T2 hyperintensity within an adrenal mass (present in 2/3 of pheochromocytomas)
- Soft-tissue density adrenal mass with avid homogeneous or heterogeneous enhancement on CT
- Areas of internal necrosis, hemorrhage, or cystic change causing heterogenous signal characteristics
- Absence of signal loss on out-of-phase imaging (no intracellular lipid content)
Key Image Findings
- On CT, pheochromocytoma demonstrates a soft-tissue density mass with overall sensitivity >95% for lesions larger than 5-10mm, and may appear heterogenous in larger tumors due to necrosis and cystic change.
- Pheochromocytoma shows avid enhancement on contrast-enhanced CT with possible 'washout' phenomenon that can mimic adrenocortical adenoma.
- On MRI, pheochromocytoma appears slightly T1 hypointense compared to normal adrenal tissue and demonstrates marked T2 hyperintensity in approximately two-thirds of cases.
- Internal hemorrhage and necrosis produce variable MRI signal characteristics depending on the age of blood products, ranging from T1 hyperintense (methemoglobin) to T1 hypointense (hemosiderin).
- Pheochromocytoma shows hyperintensity on gadolinium-enhanced images reflecting the highly vascularized nature of the tumor tissue.
- There is no appreciable signal loss on out-of-phase imaging because pheochromocytomas rarely contain significant intracellular lipids, distinguishing them from adrenocortical adenomas.
- Approximately 85-90% of pheochromocytomas arise from the adrenal gland, while 10-15% are extra-adrenal, found in the abdominal paraaortic region, Organ of Zuckerkandl, bladder, skull base, neck, or pelvis.
- Iobenguane (MIBG) I-123 scintigraphy is useful for detecting pheochromocytoma when CT or MRI is negative and for localizing extra-adrenal masses or metastases.
Differential Diagnosis
- Adrenocortical adenoma: distinguished by presence of intracellular lipids causing signal loss on out-of-phase MRI imaging and lower T2 signal intensity.
- Adrenal carcinoma: typically larger at presentation (>4cm), more heterogenous, with invasion of surrounding structures and higher likelihood of metastases.
- The tumors are said to follow a 10% rule:
- ~10% are extra-adrenal
- ~10% are bilateral
- ~10% are malignant
- ~10% are found in children
- ~10% are not associated with hypertension
- ~10% contain calcification
- Adrenal metastasis: usually multiple, bilateral, or in context of known primary malignancy; typically lower T2 signal intensity than pheochromocytoma.
- Renal clear cell carcinoma: arises from kidney rather than adrenal gland and shows different enhancement pattern with delayed washout.
- Cystic lesions/hemorrhagic cysts: lack solid enhancing component and avid enhancement characteristic of pheochromocytoma.
- Imaging artifact or normal anatomic variants: confirm with multiple imaging sequences and cross-sectional imaging modalities.
Discussion
Pheochromocytoma has an annual incidence of 0.6 per 100,000 persons, but approximately 50% are diagnosed at autopsy, indicating underdiagnosis during life.
Up to 40% of pheochromocytomas are associated with hereditary syndromes including von Hippel-Lindau syndrome, multiple endocrine neoplasia type 2, and neurofibromatosis type 1, warranting genetic screening in affected patients.
Approximately 4.2-17.7% of pheochromocytomas are malignant with potential for metastatic disease, making detection of extra-adrenal lesions and surveillance critical.
The classic triad of episodic headache, diaphoresis, and tachycardia is present in only a minority of patients, so biochemical testing (urinary and plasma fractionated metanephrines/catecholamines) is essential for diagnosis.
Preoperative alpha-adrenergic blockade followed by beta-adrenergic blockade is mandatory before surgical resection to prevent intraoperative hypertensive crisis and hemodynamic instability.
Surgical complications include hemodynamic instability, arrhythmia, renal failure, and injury to adjacent structures such as the spleen, kidney, and major vessels due to mass adhesions.
Reporting Pearls
Describe pheochromocytoma as a T1 hypointense, markedly T2 hyperintense adrenal mass with avid gadolinium enhancement and internal areas of necrosis/hemorrhage; note absence of signal loss on out-of-phase imaging to exclude adenoma, and specify location (adrenal vs. extra-adrenal) and size to guide surgical planning.
Pitfalls
- Confusing pheochromocytoma with adrenocortical adenoma based solely on enhancement pattern or size; carefully evaluate out-of-phase imaging, which shows signal loss in adenomas but not in pheochromocytomas.
- Failing to search for extra-adrenal lesions in the paraaortic region, Organ of Zuckerkandl, bladder, and other sites, missing 10-15% of tumors that are extra-adrenal.
- Misinterpreting the 'washout' enhancement pattern of pheochromocytoma on delayed CT imaging as typical of adenoma; correlation with biochemical markers and MRI signal characteristics is essential.
- Overlooking metastatic disease or malignant potential; approximately 4-18% of pheochromocytomas are malignant, and surveillance imaging should be performed to detect recurrence or metastases post-resection.